Abstract
Background
Cystic fibrosis transmembrane conductance regulator (CFTR) triple modulator therapy, elexacaftor–tezacaftor–ivacaftor (ETI) has transformed care for people with cystic fibrosis (CF) who have eligible mutations. It is, however, not curative. Response to treatment also varies and lung disease, although slowed, remains progressive. We have previously demonstrated inhibition of the epithelial sodium channel (ENaC) by selective furin inhibition to be an alternative, mutation-agnostic approach that can enhance airways hydration and restore mucociliary transport (MCT) in CF. Inhibition of furin therefore, offers a potential therapeutic strategy for those ineligible, intolerant or nonresponsive to ETI and may provide a further opportunity for clinical benefit for those currently treated with ETI. The aim of this study was to determine the impact of furin inhibition on ETI responses to assess its utility as an adjunct therapy.
Methods
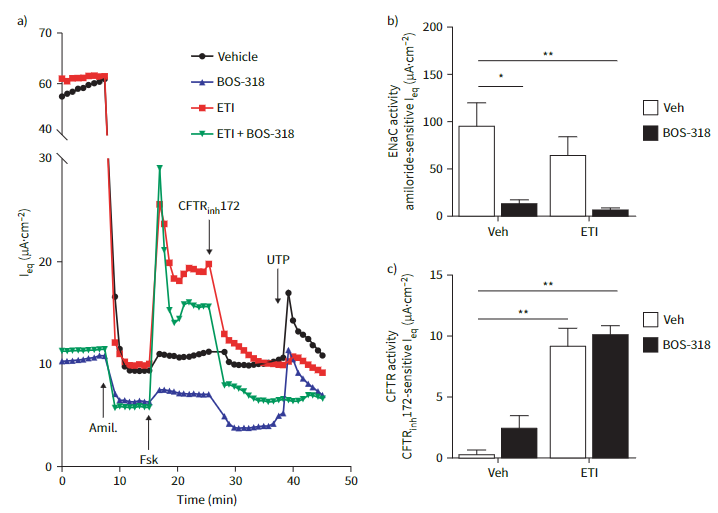
Differentiated primary CF human bronchial epithelial cells (HBECs) were treated with the highly selective furin inhibitor BOS-318 and with ETI. Ion channel function was measured using a 24-channel Transepithelial Current Clamp (TECC-24) system and airways surface hydration was investigated by measuring airway surface liquid (ASL) height and MCT rate.
Results
The presence of BOS-318 had no effect on the ability of ETI to stimulate CFTR-mediated Cl? secretion but contributed a reduced Na+ transport via robust inhibition of ENaC. This altered ion transport profile effected an improved ASL height and MCT rate, which were significantly greater than improvements observed with ETI alone, demonstrating the benefits of the dual approach.
Conclusions
Selective furin inhibition has the potential to further improve clinical outcomes for all people with CF and offers opportunity as an adjunct to improve responses to currently available CFTR modulator therapies.